





























本期国际药事新闻呈现两条并行主线:
一是监管层面从“规则制定”向“强化执行与可操作化”转变——以广告审查、现场检查替代工具、以及对质量体系缺陷的警告为代表;
二是监管与行业在“制度适配与能力建设”上同步推进——包括医疗器械法规的定向修订征询、上市后/安全研究的方法学国际趋同、以及行业大会和技术讨论推动实务路径。
一、美国FDA:监管策略改变与新药批准
1. 卫生部报告引发的监管策略改变与广告整治
上周,美国卫生与公共服务部(HHS)秘书罗伯特·F·肯尼迪(Robert F. Kennedy Jr.)公布“Make Our Children Healthy Again”(MAHA)战略报告,提出通过纠正饮食、化学暴露、缺乏运动、慢性压力与医疗过度等因素来遏制儿童慢性病。报告将多项任务委派给FDA,包括加速某些一期(Phase I)试验用药更快到患者手中、提高用户收费谈判透明度,以及要求FDA与其他部门加强对DTC药物广告的监督。
随后,FDA公开宣布针对所谓“误导性”处方药广告展开整治,计划停止长期允许广告通过所谓“adequate provision”(在广告中只作主要风险陈述并在别处提供完整标签)来规避在广告中完整披露风险的做法,并已对大量企业发出警告信与约100封“停止并终止”(cease-and-desist)类信函,其中对AstraZeneca的Flumist广告发出未命名信,指出其电视广告中风险陈述过快且视觉干扰过强,导致风险信息未被受众充分理解。
整体来看,监管话语由过去的“引导合规”明显走向“以可察觉的执法行动迫使行业改变宣传策略”,对市场准入后的商业沟通与合规治理提出更高即时性要求。

图:FDA 严厉打击虚假药品广告
2. 以“替代工具”推进检验现代化:FDA发布关于替代检查工具的最终指南
FDA本周发布了题为“替代工具:评估待审申请中的药品生产设施”的最终指南,正式把远程监管评估(Remote Regulatory Assessments, RRA)、远程交互式评估(Remote Interactive Evaluations, RIE)、以及基于与外国监管伙伴信息交换(MRA)等方式纳入可用工具清单,适用于NDA/ANDA/BLA等在审申请涉及的生产场所的 PAI/PLI(批准前/许可前检查)。最终稿在响应业界反馈后增加了对记录请求回应时间的示例(例如常规情形下15个工作日、需要译文时30个工作日等)、明确适用于境内外设施,并就不配合可能延长审评决策作出提示。对企业而言,这意味着在面对跨境供应链时需更早准备数字化文件与远程支持机制,且拒绝配合的后果将直接体现在审评时效上。
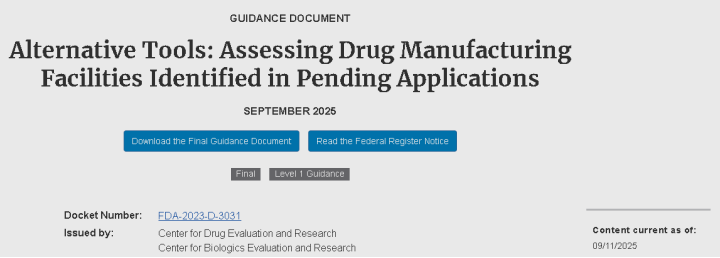
图:FDA 发布关于替代检查工具的最终指南
3. 执法关注:FDA对印度原料药企业Hikal发出警告信
FDA向印度Hikal Limited发布了针对其API生产厂的警告信,指出其质量单位(Quality Unit)未能充分调查并解决自2020年以来关于金属颗粒污染的约22起投诉,未能提供可信的根本原因分析。该警告信是FDA对跨境原料药质量监管加强执法的又一体现,提醒全球供应链上游企业在供应商管理、清洁/验证与投诉调查方面必须提供有力证据链。

图:FDA 对印度原料药企业 Hikal 发出警告信
4. 新药与新适应证批准情况
本周FDA批准了数项临床与罕见病/肿瘤领域的重要进展:
- 阿斯利康(AstraZeneca)的Koselugo(selumetinib,颗粒剂/胶囊)将适应证扩展到1岁及以上儿童,用于无法手术切除且有症状的神经纤维瘤病1型(NF1)相关的plexiform neurofibromas;
- 武田(Takeda )的VONVENDI(von Willebrand factor, recombinant)通过sBLA扩展适应证,用于成人常规预防性用药以减少出血频次,并涵盖儿科围手术及按需止血管理;
- 强生(J&J)的膀胱内给药系统Inlexzo(原名 TAR-200,gemcitabine intravesical system)被批准用于BCG无效的非肌层浸润性膀胱癌(含原位癌),为不能或拒绝根治性切除的患者提供了器官保留治疗的新选择。
上述批准体现监管在儿童用药、血液病长期管理及局部给药递送系统上的审评进展。 (U.S. Food and Drug Administration)

图:FDA新药与新适应证批准情况
二、美国PDA:微生物连续监测的技术讨论
美国注射剂协会PDA在其会刊刊登文章,讨论了在Grade A/B无菌环境实施连续微生物监测时是否应将结果规范化为CFU/m³的问题。文章指出:微生物在时间和空间上分布非均一、CFU是不可分割的整数单位,因此在持续采样且采样体积超过1 m³时,单纯归一化到1 m³可能掩盖采样期间的瞬时暴发特征;建议以“绝对 CFU + 采样体积”并辅以解释性注释和趋势分析为主,归一化值仅作为调查用途参考。该技术讨论对应EU GMP Annex 1的连续监测要求,提示企业在监测数据报告与偏差调查策略上应采用更具可解释性的呈现方式。

图:美国PDA有关微生物连续监测的技术讨论
三、欧盟EMA:医疗器械法规征询意见与制造业回流
1. 欧盟就MDR/IVDR进行“定向修订”征询(Call for Evidence)
欧盟委员会于2025年9月发起针对医疗器械法规(MDR)与体外诊断法规(IVDR)的“定向修订”征询(Call for Evidence),征求利益相关方意见(截至2025年10月6日)。官方目标为“在保持高水平公共卫生与患者安全的前提下,简化流程、降低中小企业负担、提高认证可预测性与成本效益,并为低/中风险器械提供更为相称的符合性评估路径”。该征询明显回应过去两年关于通知机构容量、行政负担与供应链稳定性的行业批评,短期内将为拟议修订的工作文件提供证据基础。

图(部分):欧盟就MDR/IVDR 进行“定向修订”征询
2. 制造回流(reshoring)与环境保护的矛盾:欧洲议会委员会辩论显现权衡
在围绕“关键药品法案”(Critical Medicines Act)提交至欧洲议会环境、气候与食品安全委员会的讨论中,部分议员(例如立陶宛)强调将生产回流与公共健康优先放在同等重要的位置,呼吁减少对生产的额外负担以吸引制造回流;另一些议员(如卢森堡、德国)则警惕把回流作为弱化环境保护标准的借口,要求在推动产业回流时不得降低现有环境法规的门槛。该讨论提醒立法者在产业政策与环境守护之间寻找制度性平衡点,并可能影响对生产许可、废水治理与环境合规许可程序的修订方向。

图:欧洲议会委员会有关药品制造业回流的辩论,显现权衡
四、英国MHRA:发布医疗器械上市后监测报告(PMSR)的标准化格式
为落实2024年修订的《医疗器械(上市后监测要求)条例》,英国药监MHRA发布了医疗器械“上市后监测要求(PMSR)”的标准化格式与填写指引,要求制造商在器械上市后三年内首次准备PMSR,并至少每三年更新一次。该标准化模板包含封面、执行摘要、设备范畴说明、性能信息(包括严重事故、召回与其他相关PMS数据)、预防和纠正措施、结论与行动计划等模块。MHRA允许在不适用的章节中留空或用其它合适形式展示数据,但要求对任何偏离模板的做法作出充分理由说明。对在英经营的器械企业而言,这一统一格式可以提高监管沟通效率,但也要求企业在PMS数据治理、信号检测与法规回应能力上做出制度化升级。

图:发布医疗器械上市后监测报告标准化格式
五、欧洲药典EDQM:聚焦药品质量安全
EDQM发布会议信息,邀请关注“将健康作为人权”的跨部门讨论,该会议将于2025年10月15日在法国斯特拉斯堡及线上举行,第3场讨论专门聚焦药品与医疗保健的质量与安全(由EDQM主任Petra Doerr主持),并汇聚EMA、WHO、患者代表、法官与执法机构等多方声音探讨打击伪劣药品、青少年患者期望变化及监管协作对策。该类会议强调了药品质量治理与人权、司法与跨机构合作之间的日益交织,提示监管机关在制定技术规则时须兼顾伦理与法律框架。
图:欧洲药典EDQM会议聚焦药品质量安全
六、ICH:M14指南进入第5步,RWD/RWE在药物安全研究中的国际方法学趋同量体系)
国际协调会议(ICH)宣布M14指南已于2025年9月完成第4步(即拟定文本的最终化阶段),目前已进入实施阶段,即第5步。指南已主题为“利用真实世界数据(RWD)进行药事流行病学研究的总体原则”,旨在为药品上市后以RWD开展非干预性药事流行病学研究的计划、设计、分析与报告提供国际统一的技术框架。该文件有望减少各地区重复研究、提升跨境提交的可接受性,并为药物安全信号评估与监管决策提供标准化方法学基础。对药企与监管科学家而言,M14的推进意味着未来安全性研究设计、数据可接受性标准与阐释路径将更趋国际一致。

图:ICH 公告
七、ISPE:2025年会,主题聚焦Pharma 4.0与实务落地
ISPE(国际制药工程学会)宣布其2025年年会与博览会(Annual Meeting & Expo)将于2025年10月26–29日在美国北卡罗来纳州夏洛特举行,议程强调数字化转型(Pharma 4.0™)、供应链韧性、监管框架与合规实务,并包含大量互动式工作坊与青年人才黑客松、女性行业网络等活动。该会为工程、质量、法规与运营各职能搭建交流平台,预期将在数字化与合规对接、AI/自动化在生产中的实际应用,以及跨部门人才培养等方面提供可操作案例。

图:美国ISPE 2025 年会,主题聚焦 Pharma 4.0与实务落地
结 语
过去一周的新闻反映出一个清晰的监管态势:监管者在“规则层面”与“执行层面”同时发动——既推动法规与指南的更新以适配新技术与全球化供应链(如 ICH M14、FDA 替代检查工具、欧盟 MDR/IVDR 的定向修订),又在合规执行上显示更强的即时性与可见度(如 FDA 对 DTC 广告的大规模警告与对质量失控的警告信)。
文章来源:蒲公英Ouryao
原文链接: https://mp.weixin.qq.com/s/iB_Mh5IJojpVmCfVyY9jig













 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论