





























1.4.1本周全球TOP10创新药研发进展
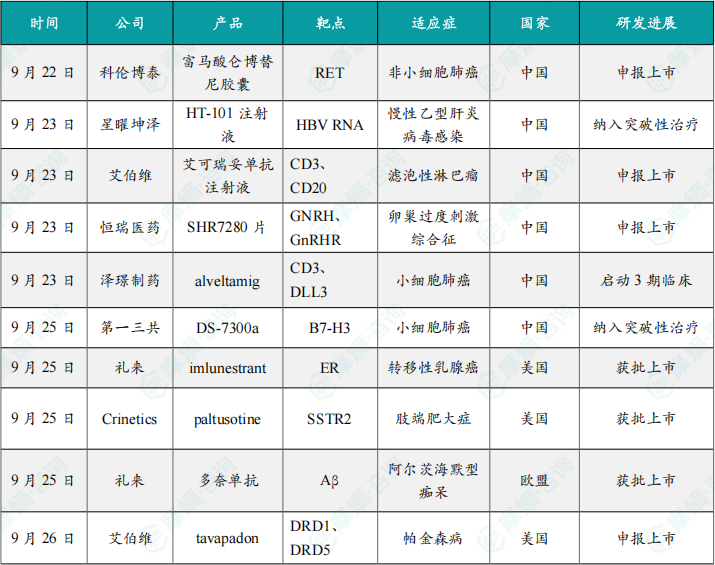
(1)科伦博泰1类新药富马酸仑博替尼申报上市
9月22日,CDE官网显示,科伦博泰1类新药富马酸仑博替尼胶囊上市申请获受理。用于治疗RET融合阳性局部晚期或转移性非小细胞肺癌(NSCLC)成人患者。这标志着我国在肺癌精准治疗领域又迈出了坚实的一步。
仑博替尼是科伦博泰自主研发的第二代小分子选择性RET抑制剂,对常见的RET基因融合和突变具有广泛活性。作为一款新型下一代选择性RET抑制剂,该药物被定位为治疗NSCLC、甲状腺髓样瘤(MTC)以及其他RET变异的高患病率实体瘤的精准治疗药物。
临床前研究数据显示,仑博替尼在体外和体内对主要RET激酶表现出良好的抑制活性,在动物模型中也展现出优异的血脑屏障穿透能力,这为其治疗脑转移患者提供了理论基础。值得注意的是,该药物具有克服第一代RET抑制剂耐药的潜力,为临床治疗提供了新的解决方案。
(2)星曜坤泽乙肝联合疗法被NMPA纳入突破性治疗品种
9月23日,星曜坤泽宣布其自主研发的HT-101注射液联合HT-102注射液正式被中国国家药监局(NMPA)纳入突破性治疗品种,拟定适应症为治疗慢性乙型肝炎病毒感染。
HT-101是一款GalNAc偶联的siRNA创新药物,通过高特异性肝脏靶向,沉默乙肝病毒(HBV)的mRNA,阻断多个病毒蛋白合成,达到抗HBV作用。
HT-102是针对HBsAg的全人源单克隆抗体,可阻止HBV再感染,清除体内循环中的含有HBsAg的亚病毒颗粒,减少亚病毒颗粒介导的免疫抑制。
HT-101注射液联合HT-102注射液理论上可同时针对外周血的HBsAg和靶向肝脏内多种HBV病毒蛋白,机制上互补,药效有潜在的协同作用。
根据星曜坤泽新闻稿介绍,此次突破性疗法认定认定基于早期临床试验中展现出的显著抗病毒活性和良好安全性。HT-101的1b期临床试验显示,所有剂量组患者在两次给药后HBsAg水平持续下降,高剂量组部分患者实现HBsAg清除。
(3)艾伯维CD3/CD20双抗艾可瑞妥单抗注射液新适应症在华申报上市
9月23日,CDE网站显示,艾伯维的艾可瑞妥单抗注射液(epcoritamab)在华申报新适应症。根据优先审评进展,本次申报的适应症为联合利妥昔单抗和来那度胺治疗复发或难治性滤泡性淋巴瘤(FL)成人患者。
艾可瑞妥单抗是Genmab利用其专有的DuoBody技术开发的一款IgG1双特异性抗体,可同时靶向T细胞上的CD3和B细胞上的CD20,诱导T细胞杀伤CD20+细胞。
2020年6月,艾伯维与Genmab达成协议,共同开发和商业化包括该产品在内的3款双抗。8月7日, 一项评估艾可瑞妥单抗联合利妥昔单抗+来那度胺对比利妥昔单抗+来那度胺方案,用于成人复发或难治性滤泡性淋巴瘤(R/R FL)患者的疗效与安全性的III期研究(EPCORE FL-1研究)取得积极结果。预设中期分析显示,该研究已达到ORR和PFS双重主要终点。
2023年5月,艾可瑞妥单抗首次在美国获批上市,用于治疗接受过二线或多线系统治疗的复发或难治性弥漫性大B细胞淋巴瘤(R/R DLBCL)患者。2024年6月,FDA又批准该药用于治疗至少接受过两种系统治疗的复发或难治性滤泡性淋巴瘤患者。
(4)恒瑞创新药SHR7280片用于辅助生殖适应症上市申请获受理
9月23日,恒瑞医药发布公告称,其自主研发的1类创新药SHR7280片的药品上市许可申请获受理。该药物适应症为:在辅助生殖技术中,用于控制性卵巢刺激治疗的患者,预防早发黄体生成素(LH)峰。
此次申报上市,是基于一项在不孕症女性受试者中开展的多中心、随机、双盲双模拟、阳性药物对照的非劣效性Ⅲ期临床研究(SHR7280-302)。2024年11月,该研究达到了方案预设的主要研究终点。该研究由北京大学第三医院乔杰院士担任主要研究者,共有22家国内中心参与本研究,共入组317例不孕症女性受试者。研究结果表明:在辅助生殖技术控制性卵巢刺激治疗中,口服SHR7280片与皮下注射醋酸加尼瑞克注射液具有相当的临床疗效,SHR7280可有效预防早发LH峰,防止提前排卵,且整体安全性良好。
继2023年恒瑞医药与默克就PARP1抑制剂HRS-1167达成首次合作后,2025年4月双方再度携手,恒瑞医药将SHR7280(适应症涵盖医学辅助生殖及妇科领域)在中华人民共和国大陆地区(不含香港特别行政区、澳门特别行政区和台湾地区)的独家商业化权益授予默克。
(5)泽璟制药CD3/DLL3三抗alveltamig启动3期临床
9月23日,药物临床试验登记与信息系公式平台显示,泽璟制药的CD3/DLL3三抗药物ZG006(alveltamig)启动了首个III期临床试验。据数据统计,该药物是全球首款启动III期临床的DLL3三抗药物,也是全球首款进入III期阶段的三抗药物。
该研究是一项多中心、随机对照、开放标签临床试验(n=420),旨在评估ZG006(10mg,每2周1次)对比研究者选择的化疗方案治疗复发性小细胞肺癌(SCLC)患者的有效性和安全性,研究的主要终点是总生存期(OS)。
ZG006是泽璟制药基于其双/多抗药物研发平台开发的一款靶向CD3和两个不同DLL3表位的三特异性T细胞衔接器。临床前研究结果显示,ZG006在小鼠肿瘤模型上具 有显著的肿瘤抑制作用,可以导致显著比例的小鼠肿瘤完全消退,说明ZG006具有强效的肿瘤杀伤作用。
(6)第一三共小细胞肺癌新药DS-7300a在中国拟纳入突破性治疗品种
9月25日,中国国家药监局药品审评中心(CDE)官网公示显示,第一三共(Daiichi Sankyo)申报1类新药 Ifinatamab Deruxtecan(I-DXd, DS-7300a)拟纳入突破性治疗品种,拟定适应症为:治疗在含铂化疗期间或之后出现疾病进展的广泛期小细胞肺癌成人患者。
公开资料显示,这是默沙东(MSD)与第一三共联合开发的潜在"first-in-class"的B7-H3靶向抗体偶联药物(ADC)。针对本次的适应症,I-DXd已经于今年8月被美国FDA授予突破性疗法认定。
I-DXd是一种B7-H3靶向ADC,采用第一三共专有的DXd ADC技术设计而成,基于人源化B7-H3靶向IgG1单克隆抗体构建。该抗体通过基于四肽的可裂解连接子与多个拓扑异构酶I抑制剂有效载荷相连。
(7)礼来口服SERD新药imlunestrant获FDA批准上市
9月25日,礼来宣布,FDA已批准其口服雌激素受体拮抗剂 Inluriyo (imlunestrant, 200mg片剂) 用于治疗雌激素受体阳性 (ER+)、人表皮生长因子受体2阴性 (HER2-)、携带ESR1突变的晚期或转移性乳腺癌 (MBC) 成人患者,这些患者的疾病在至少接受过一线内分泌疗法 (ET) 后出现进展。
此前,全球仅有一款口服SERD药物即艾拉司群(Menarini)获批上市,此外同类药物中 camizestrant(阿斯利康)已申报上市,giredestrant(罗氏)也于近期宣告III期研究成功。
Inluriyo适用于ER+、HER2-、携带ESR1突变的MBC。一些乳腺癌会发展出ESR1突变,这些突变可导致雌激素受体过度活跃并驱动癌症生长。Inluriyo能够结合、阻断并促进这些受体的降解,从而有助于减缓疾病进展。其每日一次的口服给药方式为患者提供了一种便捷的治疗选择。
(8)首个肢端肥大症SST2激动剂口服药获FDA批准上市
9月25日,Crinetics公司宣布,FDA已经批准新型非肽类SST2(生长抑素受体2型)激动剂paltusotine(商品名:Palsonify)作为首个肢端肥大症口服药上市。
肢端肥大症的市场长期以来由Ipsen和诺华占据,作为首个新机制口服药,此次paltusotine获得批准将意味着现有治疗格局将出现重大转变。本次获批基于两项III期临床试验PATHFNDR-1和PATHFNDR-2的数据。
PATHFNDR-1针对的是之前有注射治疗史的患者,2023年公布的结果显示服用该药物的患者中有83%达到了IGF-1水平的正常上限,而服用安慰剂的患者中只有4%达到这一水平。
PATHFNDR-2试验针对的则是之前没有接受过注射治疗的患者,2024年公布的PATHFNDR-2试验结果显示,服用Palsonify的患者中有56%达到了正常的IGF-1水平,而安慰剂组中这一比例仅为5%。
(9)礼来治疗阿尔茨海默病药物多奈单抗在欧盟获批上市
9月25日,礼来宣布,欧洲委员会(EC)已授予Kisunla(Donanemab,多奈单抗,也被简称为“D药”)上市许可,用于治疗早期症状性阿尔茨海默病(AD)成人患者,包括患有轻度认知障碍以及患有轻度痴呆阶段AD且确认存在淀粉样蛋白病理的载脂蛋白E(ApoE4)杂合子或非携带者。
礼来新闻稿指出,在III期TRAILBLAZER-ALZ 2研究中,D药在早期症状性AD患者身上展现了有意义的成果,显著减缓了认知和功能衰退。且患者越早被识别、诊断并用采取治疗,治疗效果越好。
D药基于β淀粉样蛋白(Aβ)级联假说研发,于2024年7月获FDA批准上市,是继渤健/卫材的仑卡奈单抗后,全球第二款获FDA批准上市的Aβ单抗产品。此前,D药已分别于美国、日本、中国及英国等地获批上市,但在欧盟的上市申请此前因严重的淀粉样蛋白相关影像学异常(ARIA)风险一直未被批准。
(10)艾伯维向FDA递交帕金森小分子疗法上市申请
9月26日,艾伯维(AbbVie)宣布已向美国FDA提交在研小分子 tavapadon 的新药申请(NDA),用于帕金森病治疗。
Tavapadon是一种新型选择性多巴胺D1/D5受体部分激动剂,被开发为每日一次口服疗法。申请主要基于TEMPO临床开发项目的结果,该项目全面评估了tavapadon在帕金森病不同患者群体中的疗效、安全性及耐受性。
TEMPO项目包含三项关键3期临床试验:TEMPO-1与TEMPO-2在早期帕金森病患者中开展,结果显示患者在第26周时,运动障碍协会-统一帕金森病评定量表(MDS-UPDRS)第2和3部分综合评分上,与基线相较具有统计学显著改善。TEMPO-3试验则评估tavapadon联合左旋多巴的疗效,结果表明患者处于“on”时期(症状得到良好控制且无运动障碍)明显延长。
1.4.2本周全球TOP10积极/失败临床结果
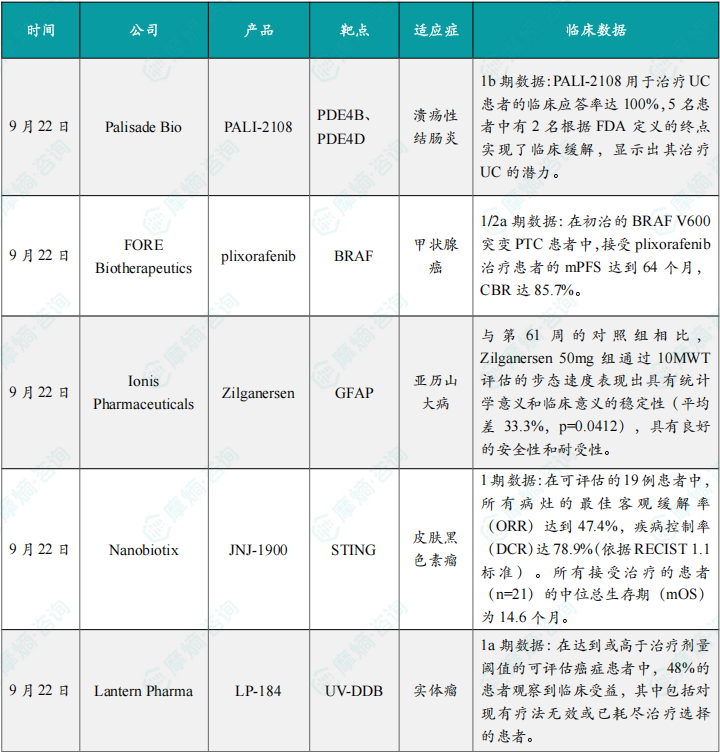
(1)Palisade Bio公司公布溃疡性结肠炎在研药物PALI-2108的1b期临床试验数据
9月22日,Palisade Bio公司公布了其用于治疗溃疡性结肠炎的在研药物 PALI-2108 的1b期临床试验数据。在UC和纤维狭窄型克罗恩病(FSCD)中,较高的PDE4B表达与局部炎症活动有关。PALI-2108 是一种PDE4抑制剂前药,能在UC和FSCD等疾病部位局部活化,显著提升病变组织内PDE4抑制剂浓度,同时减少全身暴露,从而降低腹泻等传统PDE4抑制剂的不良反应。
1a期安全性和药代动力学(PK)研究结果显示,PALI-2108 在临床试验中表现出良好的安全性,未报告严重不良事件,也无实验室指标异常或心电图异常。在1b期临床试验中,PALI-2108 用于治疗UC患者的临床应答率达100%,5名患者中有2名根据FDA定义的终点实现了临床缓解,显示出其治疗UC的潜力。生物标志物分析显示,UC患者在基线时升高的186个与纤维化及克罗恩病狭窄相关的基因表达得到正常化,表明PALI-2108在治疗FSCD方面也具有一定潜力。
(2)FORE Biotherapeutics公司公布plixorafenib 1/2a期临床试验数据
9月22日,FORE Biotherapeutics公司公布了 plixorafenib 的1/2a期临床试验的新数据。Plixorafenib 是一款可口服的下一代小分子BRAF突变体选择性抑制剂,旨在靶向广泛的BRAF突变体,同时不影响野生型RAF蛋白的活性。
此次公布的结果显示,plixorafenib 用于治疗BRAF基因变异的乳头状甲状腺癌和甲状腺未分化癌(ATC)可实现持久的疾病控制,与现有标准疗法的历史数据相比表现更为优越,且安全性良好,与既往报告的药物安全性特征一致。在初治的BRAF V600突变PTC患者中,接受plixorafenib治疗患者的mPFS达到64个月,CBR达85.7%。
(3)Ionis公布其新药Zilganersen关键研究的积极顶线结果
9月22日,反义寡核苷酸(ASO)龙头Ionis Pharmaceuticals宣布了其在研的一款亚历山大病(AxD)治疗药物——Zilganersen(ION373)关键研究的积极顶线结果。
AxD是一种罕见的、进行性且通常致命的神经系统疾病。该病由神经胶质纤维酸性蛋白(GFAP)基因中的致病变异引起,通常在症状出现后14-25年内导致死亡,目前,该疾病领域尚无批准的疾病缓解药物。
Zilganersen可阻止由GFAP基因致病变异而导致的GFAP的过度产生和累积,是第一个也是唯一一个证明对AxD具有临床意义和疾病缓解影响的药物。研究结果显示,与第61周的对照组相比,Zilganersen 50mg组通过10MWT评估的步态速度表现出具有统计学意义和临床意义的稳定性(平均差 33.3%,p=0.0412),具有良好的安全性和耐受性。此外,Zilganersen在关键次要终点中也表现出一致的益处。这些数据标志着研究药物首次显示出对AxD的积极疾病缓解作用。
(4)Nanobiotix公司公布JNJ-1900 1期联合治疗皮肤黑色素瘤最新结果
9月22日,Nanobiotix公司公布了一项正在进行的1期研究的最新结果。该研究评估了JNJ-1900联合免疫检查点抑制剂治疗晚期癌症患者的效果,此次分析重点聚焦于原发性皮肤黑色素瘤患者。
JNJ-1900由功能化二氧化铪(HfO2)纳米颗粒组成,经由一次性瘤内注射给药并通过放射疗法激活。它的物理作用机制为:通过放疗激活,诱导被注射肿瘤内大量的肿瘤细胞死亡,随后触发适应性免疫反应和长期的抗癌记忆。得益于该物理作用机制,Nanobiotix公司认为该药物可扩展到任何可以通过放疗治疗的实体肿瘤和任何联合治疗方案中,特别是与免疫检查点抑制剂联合。
2023年,Nanobiotix宣布与强生旗下杨森公司(现名为强生创新制药)达成全球共同开发和商业化该药物的许可协议。
此次公布的结果显示,在既往接受过多线治疗(包括抗PD-1治疗)后疾病进展的黑色素瘤患者中,JNJ-1900联合疗法表现出良好的安全性及早期疗效信号。在可评估的19例患者中,所有病灶的最佳客观缓解率(ORR)达到47.4%,疾病控制率(DCR)达78.9%(依据RECIST 1.1标准)。所有接受治疗的患者(n=21)的中位总生存期(mOS)为14.6个月。研究者认为,这些数据支持通过随机临床试验进一步验证该方案作为抗PD-1治疗初治或耐药原发性皮肤黑色素瘤患者的潜在新选择。
(5)Lantern Pharma公司在研疗法LP-184的1a期临床试验达到终点
9月22日,Lantern Pharma公司宣布,其在研疗法LP-184的1a期临床试验达到了所有主要终点,显示出良好的安全性和PK特征,并观察到初步的抗肿瘤活性。目前患者入组已完成,部分患者因持续获得临床获益而继续接受治疗。LP-184是一种可穿透血脑屏障的小分子药物,通过“合成致死”机制靶向DNA损伤修复(DDR)缺陷肿瘤。
此次公布的结果显示,在达到或高于治疗剂量阈值的可评估癌症患者中,48%的患者观察到临床受益,其中包括对现有疗法无效或已耗尽治疗选择的患者。值得注意的是,在胶质母细胞瘤(GBM)、胃肠道间质瘤(GIST)和胸腺癌等难治性肿瘤中观察到持久的临床获益。生物标志物分析显示,LP-184在DDR通路相关基因(如CHK2、ATM、STK11/KEAP1)突变的患者中具有显著潜力,此类患者出现了明显的肿瘤缩小。此外,该药良好的安全性和耐受性为其作为单药治疗或与PARP抑制剂、免疫疗法联合使用提供了支持。
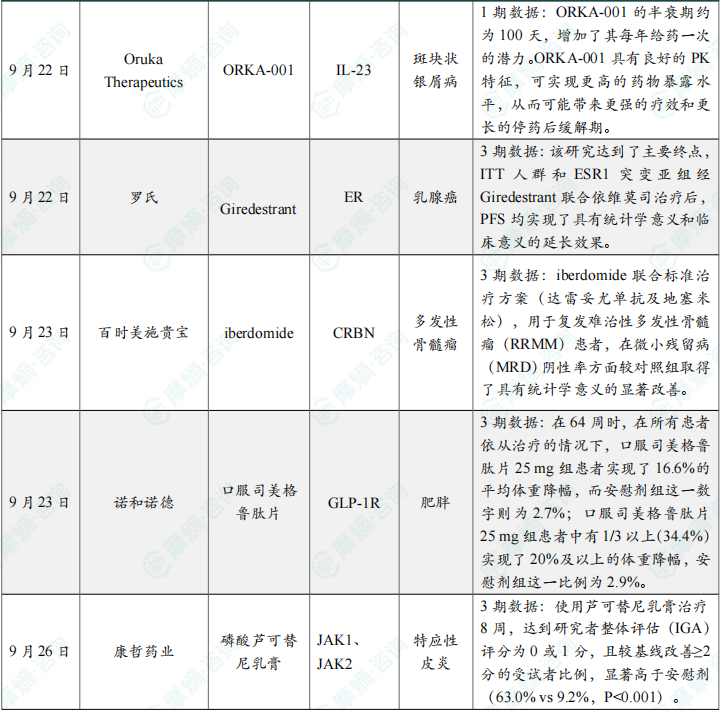
(6)Oruka Therapeutics公司公布ORKA-001 1期临床试验中期数据
9月22日,Oruka Therapeutics公司公布了其长效IL-23p19抗体ORKA-001的1期临床试验的中期数据。
ORKA-001是一种新型皮下注射的半衰期延长型单克隆抗体,靶向IL-23p19,旨在为包括斑块状银屑病(PsO)在内的慢性皮肤病提供新的治疗方法。该药物在设计上与经过验证的risankizumab具有相似的表位结合和亲和力,有望实现每年给药一到两次的给药频率。
临床数据显示,ORKA-001的半衰期约为100天,增加了其每年给药一次的潜力。ORKA-001具有良好的PK特征,可实现更高的药物暴露水平,从而可能带来更强的疗效和更长的停药后缓解期。此外,该药物在试验中耐受性良好,安全性特征与IL-23p19类药物一致。
(7)罗氏口服SERD药物Giredestrant 3期研究成功
9月22日,罗氏宣布 Giredestrant 治疗乳腺癌的III期evERA研究取得了积极结果。新闻稿指出,这是第一个在头对头标准治疗的III期研究中取得积极结果的基于口服SERD的全口服方案。罗氏计划将数据提交至监管机构,尽快使患者能够获得最新治疗选择。
Giredestrant是罗氏自主研发的一款下一代口服选择性雌激素受体降解剂(SERD)和完全拮抗剂,旨在阻止雌激素与雌激素受体(ER)结合,诱导其降解,从而阻止或减缓癌细胞的生长。
evERA研究乳腺癌是一项随机、开放标签、多中心临床试验,评估了Giredestrant联合依维莫司对比医生选择的内分泌疗法联合依维莫司治疗在辅助治疗或局部晚期/转移性环境中接受过CDK4/6抑制剂和内分泌疗法治疗的ER阳性且HER2阴性(ER+/HER2-)的局部晚期或转移性乳腺癌患者的疗效和安全性。
结果显示,该研究达到了主要终点,ITT人群和ESR1突变亚组经Giredestrant联合依维莫司治疗后,PFS均实现了具有统计学意义和临床意义的延长效果。总生存期(OS)数据尚未成熟,但显示出明显的积极趋势。后续将继续随访以进行系统分析。安全性方面,Giredestrant联合依维莫司的耐受性良好,不良事件与各自单药在既往研究中的已知安全性一致,未观察到新的安全性信号。
(8)百时美施贵宝首款CELMoD药物3期临床迎来关键胜利
9月23日,百时美施贵宝(BMS)宣布,其III期研究EXCALIBER-RRMM的预设中期分析显示,iberdomide(一种在研的cereblon E3连接酶调节剂,CELMoD)联合标准治疗方案(达雷妥尤单抗及地塞米松),用于复发难治性多发性骨髓瘤(RRMM)患者,在微小残留病(MRD)阴性率方面较对照组取得了具有统计学意义的显著改善。
根据研究设计与数据监测委员会的建议,研究将不做改变继续进行,以评估另一共同主要终点——无进展生存期(PFS),以及关键次要终点——总生存期(OS)和安全性。研究中,iberdomide与达雷妥尤单抗和地塞米松联合使用的安全性与既往研究基本一致。
iberdomide是一款分子胶蛋白质降解药物,属于新一代cereblon E3连接酶调节剂(CELMoD),通过靶向降解关键转录因子(如Aiolos和Ikaros)发挥抗癌作用。iberdomide是BMS收购Celgene获得的三种蛋白质降解药物之一,被BMS定位为Revlimid和Pomalyst等重磅血癌药物的后继者。
(9)诺和诺德口服司美格鲁肽片减重药疗效结果公布
9月23日,诺和诺德(Novo Nordisk)宣布,《新英格兰医学杂志》(The New England Journal of Medicine)近日发表了来自3期试验OASIS 4的结果。此项试验评估了在研的每日一次口服司美格鲁肽片25 mg(Wegovy片剂)的有效性和安全性,标志着诺和诺德在推进肥胖症治疗方面达成一项重要的里程碑。在此项为期64周的试验中,口服司美格鲁肽片25 mg联合生活方式干预,在307名肥胖或超重且伴有至少一种体重相关合并症但无糖尿病的成人患者中与安慰剂进行了对比。
结果显示,在64周时,在所有患者依从治疗的情况下,口服司美格鲁肽片25 mg组患者实现了16.6%的平均体重降幅,而安慰剂组这一数字则为2.7%;口服司美格鲁肽片25 mg组患者中有1/3以上(34.4%)实现了20%及以上的体重降幅,安慰剂组这一比例为2.9%。这些数字与此前进行的Wegovy注射液各项试验结果相当。
(10)康哲药业的磷酸芦可替尼乳膏特应性皮炎适应症国内3期临床达到主要终点
9月26日,康哲药业宣布,其皮肤健康业务公司德镁医药的创新药磷酸芦可替尼乳膏轻中度特应性皮炎(AD)的中国III期药物临床研究取得了积极结果。
该试验是一项在中国人群开展的随机、双盲、安慰剂对照、多中心的临床研究,共入组192例患者,旨在评估产品治疗轻中度AD的安全性和有效性。
该研究组长单位为上海市皮肤病医院,主要研究者为史玉玲教授。产品用于轻中度特应性皮炎的中国III期临床研究成功达到主要终点,即使用芦可替尼乳膏治疗8周,达到研究者整体评估(IGA)评分为0或1分,且较基线改善≥2分的受试者比例,显著高于安慰剂(63.0% vs 9.2%,P<0.001)。
关键次要终点,芦可替尼乳膏治疗8周达到湿疹面积及严重程度指数评分较基线至少改善75%(EASI 75)的受试者比例亦显著优于安慰剂(78.0% vs 15.4%,P<0.001)。
安全性方面,治疗期出现的不良事件(TEAE)的严重程度大多数为轻度或中度,未发生导致研究药物用药终止的治疗期出现的不良事件(TEAE),整体安全耐受性良好。
同期事件:
1. 2025年第39周09.22-09.28国内创新药/改良型新药申请临床/获批临床/申请上市/获批上市数据分析
2. 2025年第39周09.22-09.28国内仿制药/生物类似物申报/审批数据分析
3. 2025年第39周09.22-09.28国内医药大健康行业政策法规汇总
以上内容均来自{ 摩熵咨询医药行业观察周报(2025.09.22-2025.09.28) },如需查看或下载完整版报告,可点击!
想要解锁更多药物研发信息吗?查询摩熵医药(原药融云)数据库(vip.pharnexcloud.com/?zmt-mhwz)掌握药物基本信息、市场竞争格局、销售情况与各维度分析、药企研发进展、临床试验情况、申报审批情况、各国上市情况、最新市场动态、市场规模与前景等,以及帮助企业抉择可否投入时提供数据参考!注册立享15天免费试用!
















 浙公网安备33011002015279
浙公网安备33011002015279 本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
本网站未发布麻醉药品、精神药品、医疗用毒性药品、放射性药品、戒毒药品和医疗机构制剂的产品信息
收藏
登录后参与评论
暂无评论